
Molecular Biology: From DNA to RNA to Protein
1 Protein Structure and Function

Learning Objectives
When you have mastered the information in this chapter, you should be able to:
Levels 1 and 2 (Knowledge and Comprehension)
- Draw and label the chemical structure of single amino acid, a dipeptide, or a tripeptide and identify the various components and bonds (especially peptide bonds)
- Classify the amino acid side chains as polar and charged, polar and uncharged, hydrophobic, or special.
- Predict the types of non-covalent interactions possible between any two amino acids.
- Compare and contrast the different levels or orders of protein structure and how they relate to one another.
- Differentiate between -sheet, -helix, and ‘random coil’ structures based on the atomic interactions involved in each.
- Describe the biochemical information that determines the final three- dimensional structure and explain what powers the formation of this structure
 Level Up (Application, Analysis, Synthesis)
Level Up (Application, Analysis, Synthesis)
- Explain why the amino acid glycine is a disruptor of alpha-helical polypeptide structure.
- Identify the role of disulfide bonds in protein folding, and explain why secreted proteins often contain disulfide bonds.
- Predict how given amino acid substitutions may affect the structure and possible function of a protein.
Before you begin make sure you are thinking about answers to the learning objectives. Level 1 and Level 2 form the foundation. Level Up will be the target for assessments.
1.1 Introduction: From Computer to the Clinic and Beyond
“Imagine that you are a scientist probing the secrets of living systems not with a scalpel or microscope, but much deeper — at the level of single molecules, the building blocks of life.
You’ll focus on the detailed, three-dimensional structure of biological molecules. You’ll create intricate models of these molecules using sophisticated computer graphics. You may be the first person to see the shape protein offers clues about the role it plays in the body. It may also hold the key to developing new medicines, materials, or diagnostic procedures. You are part of the growing field of structural biology.” (1)
The molecules whose shapes most tantalize structural biologists are proteins. Virtually everything that goes on inside of cells happens as a result of the actions of proteins. Nature has programmed proteins to do nearly every job in the body:
- protein enzymes catalyze the vast majority of cellular reactions,
- proteins mediate signaling,
- give structure to individual cells and to multicellular organisms and
- proteins exert control over the expression of genes.
Life, as we know it, would not exist if there were no proteins. Therefore, it is not surprising that diseases at the molecular level are often due to the malfunction of these proteins.
Like many everyday objects, proteins are shaped to get their job done. The shape or structure of a protein offers clues about the role it plays in the body. It also holds the key to developing new medicines, materials, or diagnostic procedures. As a result, efforts to solve, predict and modify protein structures have been central to therapeutic science and the development of lifesaving and life-altering medicines for often debilitating symptoms.
Some examples include injectable insulin, antiretroviral therapy for AIDs, Celebrex a drug to treat arthritis to name just a few.
There are a variety of experimental techniques for solving protein structures- nuclear magnetic resonance, X-ray crystallography, and cryo-electron microscopy. However, to date, scientists only know the structure of a tiny fraction of all the known proteins. For long scientists have grappled with 2 research problems.
- The “protein folding problem” – the general problem of predicting protein structure directly from the amino acid sequence.
- The “protein design problem”- starting with the desired shape – something brand new and finding the amino acid sequence that can adopt that shape.
In recent years there has been a revolution in computational and AI methods for both predicting how proteins fold based on knowledge of their amino acid sequence AND creating new designer proteins from scratch!
Watch the following TED talk by Dr. David Baker, named as one of the world’s most influential scientists talks about his work, and why it’s important to create new proteins.
Molecular Biology in the News: “Artisanal Proteins”
WATCH: TED Talk by Dr. David Baker.
Link here: https://youtu.be/PJLT0cAPNfs or directly embedded below.
In order to understand how scientists can design proteins OR predict the structure of proteins from a given amino acid sequence, we need to understand the rules of protein folding.
In this chapter, we first dive into how a protein adopts its shape. We then look at X-ray crystallography one of the methods for obtaining atomic level information about protein shape.
In Chapter 2 you will learn how one can exploit the differences in proteins to separate a given protein from a complex mixture inside cells.
First, let’s review what proteins are made of.
1.2 Building Blocks of Proteins
The building blocks of all proteins are amino acids. The sequence of amino acids in individual proteins is encoded in the DNA of the cell. All amino acids have the same basic structure, which is shown in Figure 1.1
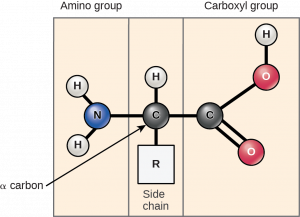
At the “center” of each amino acid is a carbon called the α-carbon and attached to it are four groups – hydrogen, an α-carboxyl group, an α-amine group, and an R-group, often referred to as a side chain. The α-carbon, carboxyl, and amino groups are common to all amino acids, so the R-group is the only unique feature in each amino acid. (A minor exception to this structure is that of proline, in which the end of the R-group is attached to the α-amine.)
With the exception of glycine, which has an R-group consisting of a hydrogen atom, all of the amino acids in proteins have four different groups attached to them and consequently can exist in two mirror-image forms, L and D. With only very minor exceptions, every amino acid found in cells and in proteins is in the L configuration.
Cells use only 20 amino acids to make polypeptides and proteins (these are specified by the genetic code), although they do use a few additional amino acids for other purposes.
The 20 amino acids have diverse properties which are determined by the chemistry of the side chains.
NOTE: If you compare groupings of amino acids in different textbooks, you will see different names for the categories. Because there are some amino acids that cannot neatly be categorized into one or another you may also see sometimes the same amino acid being categorized differently by different authors, depending on where you look.
One grouping is shown in Figure 1.2 below.

Note that some amino acids have side chains that can be ionized. They can give up a proton or take a proton. This ability is dependent on the pH! The amino acids in the figure are shown at physiological pH 7.0.
Classifying amino acids by properties is important to learn how to do- because the property affects which non-covalent interactions amino acids can form- which in turn determines protein shape.
For our purposes we will broadly group them into Acidic, Basic, Polar Neutral, and Non-polar Neutral.
TRY THIS
You should be able to infer the properties of the side chain from the 2D chemical diagram and the 3D structure. For example, which amino acids have polar side chains? Which have planar aromatic groups?
You can do this without memorizing!
- When given a structural formula for an amino acid, you can determine its type by asking three simple questions!
First: Study the different groups of amino acids shown in Figure 1-2. We want to classify them as Acidic, Basic, Polar Neutral, and Non-polar Neutral. What features did you look for?
Come up with the 3 simple “yes or no” questions you could ask based on your analysis to plug into the flowchart.
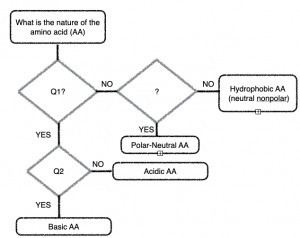
Hint: Think about the chemistry of acids and bases (what do the acidic side chains look like?), Think about what makes a covalent bond a ‘polar covalent bond?
1.3 Levels (Orders) of Structure
Now that we are familiar with the building blocks of proteins, we dive into how strings of amino acids twist and buckle, folding in upon themselves, the knobs of some amino acids nestling into grooves in others.
We shall examine protein structure at four distinct levels (Figure 1.3)
1) how the sequence of the amino acids in a protein (primary structure) gives identity and characteristics to a protein;
2) how local interactions between one part of the polypeptide backbone and another affect protein shape (secondary structure);
3) how the polypeptide chain of a protein can fold to allow amino acids to interact with each other that are not close in primary structure (tertiary structure); and
4) how different polypeptide chains interact with each other within a multi-subunit protein (quaternary structure)
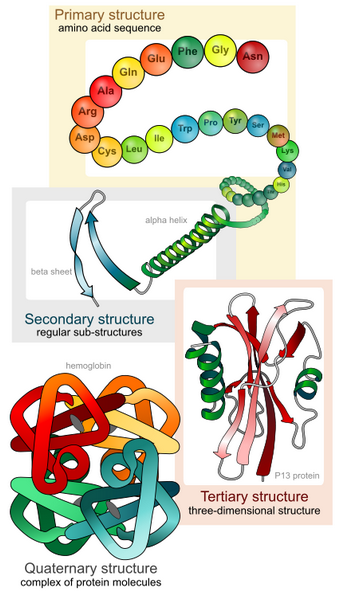
It is important to note that for each polypeptide the final structure (native fold) is the tertiary structure.
Quaternary structure refers to associations of two or more polypeptides, creating higher-order protein structures. Superimposed on these basic levels are other features of protein structure. These are created by the specific amino acid configurations in the mature, biologically active protein.
1.3.1 Primary Structure
The specific order of amino acids in a protein is known as its primary structure. Inside a cell synthesis of proteins occurs in the ribosomes and proceeds by joining the carboxyl terminus of the first amino acid to the amino terminus of the next one (Figure 1.4).
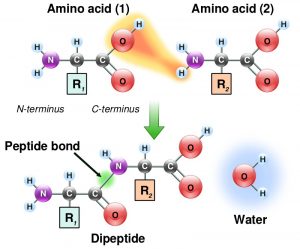
Note the chemistry of the reaction! Individual amino acids are joined together by the attack of the nitrogen of an amino group of one amino acid on the carbonyl carbon of the carboxyl group of another to create a covalent peptide bond and yield a molecule of water.
This is a condensation reaction or dehydration reaction that is common in the making of all biological polymers.
Amino acids that are part of a protein are referred to as ‘amino acid residue(s)’. You will read and hear this term being utilized often.
Because the synthesis takes place from the alpha-amino group of one amino acid to the carboxyl group of another amino acid, the result is that there will always be a free amino group on one end of the growing polymer (the amino or N-terminus) and a free carboxyl group on the other end (the carboxyl or C-terminus).
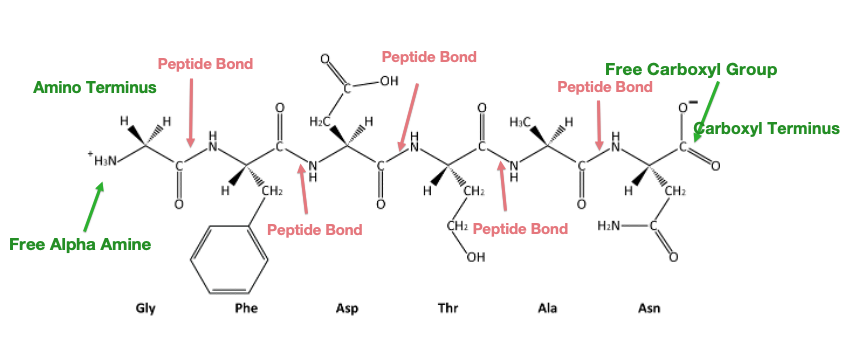
Since proteins are synthesized starting with the amino terminus and ending at the carboxyl terminus, therefore by convention amino acid sequences are written left to right from amino to carboxy-terminus. The name of the N-terminal residue is always the first amino acid. The name of each amino acid then follows.
Reversing the directionality indicates a very different protein sequence!
It is the order of amino acids that dictates the 3-D conformation (shape) the folded protein will have. This conformation, in turn, will determine the function of the protein.
Practice Exercises
1.3.2 Secondary Structure
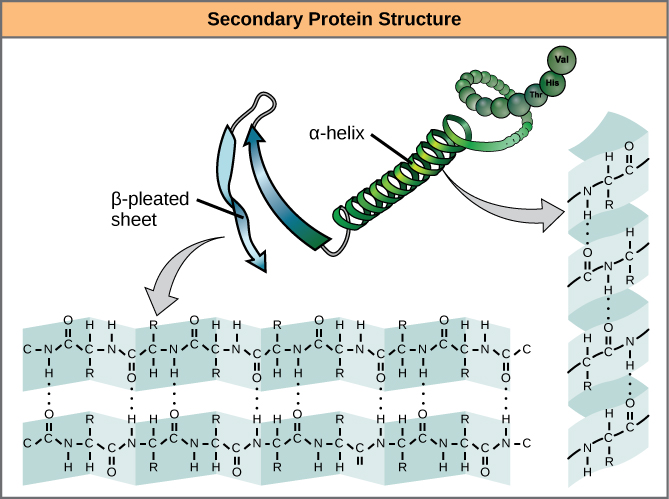
As protein synthesis progresses, interactions between amino acids close to each other begin to occur, giving rise to local patterns called secondary structures.
Two common types of secondary structures in proteins are alpha (α) helices and beta (β) strands/sheets. (Figure 1.6)
Secondary structure conformations occur due to the spontaneous formation of hydrogen bonds between amino groups and oxygens along the polypeptide backbone.
α-helix
In the α-helix, hydrogen bonds form between C=O groups and N-H groups within the polypeptide backbone that is four amino acids distant. These hydrogen bonds are the primary forces stabilizing the α-helix.
The cartoon representation of a protein’s structure represents α-helices as coils.
β strand versus β-sheet
A flattened form of the helix in two dimensions is a common description for a β- strand. Rather than coils, β-strands have bends and these are sometimes referred to as pleats, like the pleats in a curtain.
Stretches of β-strands embedded within a single polypeptide chain form a β-sheet. Within a β-sheet, hydrogen bonds form between the backbone atoms of separate β-strands. When multiple strands from different regions of a polypeptide interact in this way a relatively flat, sheet-like surface is created, the β-sheet.
The cartoon representation of a protein’s structure represents β-sheets as flat arrows.
Segments of the peptide backbone that do not form secondary structures are called loops. Loops are linkers that connect regions of secondary structure. Loops are shown in cartoon representations as lines connecting regions of secondary structure.
1.3.3 Tertiary Structure
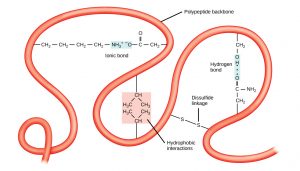
For all proteins, the unique final three-dimensional structure adopted by the polypeptide is its tertiary structure.
This structure is determined by all types of non-covalent interactions that involve amino acid side chains (side-chain and backbone interactions, or side-chain-side chain interactions).
These non-covalent bonds can be
Ionic interactions (strongest): between pairs of charged amino acids also known as salt bridges.
Hydrogen bonds: between polar groups- one of these polar groups is acting as a hydrogen donor the other as a hydrogen acceptor.
van-der Waals interactions (weakest): act only over short distances although they are present between any pair of atoms in close proximity.
Further, proteins fold in an aqueous environment, and that environment is critical to how the proteins adopt their native conformation.
Hydrophobic molecules do not form strong bonds with water. In aqueous solutions, hydrophobic molecules are driven together to the exclusion of water. As a protein folds to its final three-dimensional structure, the hydrophobic parts of the protein are forced together and away from the aqueous environment of the cell.
Amino acid residues involved in these interactions can come from distant parts of the polypeptide chain bringing the chain into a more compact shape.
Disulfide bonds stabilize tertiary structure
Based on non-covalent bonds, tertiary structures are nonetheless strong simply because of the large numbers of otherwise weak interactions that form them.
However, one covalent bond is possible and is formed when two cysteine amino acids (sulfhydryl-containing side chains) are close together as a result of tertiary structure formation.
The sulfhydryl group is highly reactive and will covalently bond with another sulfhydryl group to form a covalent disulfide bond (the disulfide (–S-S-) bond).
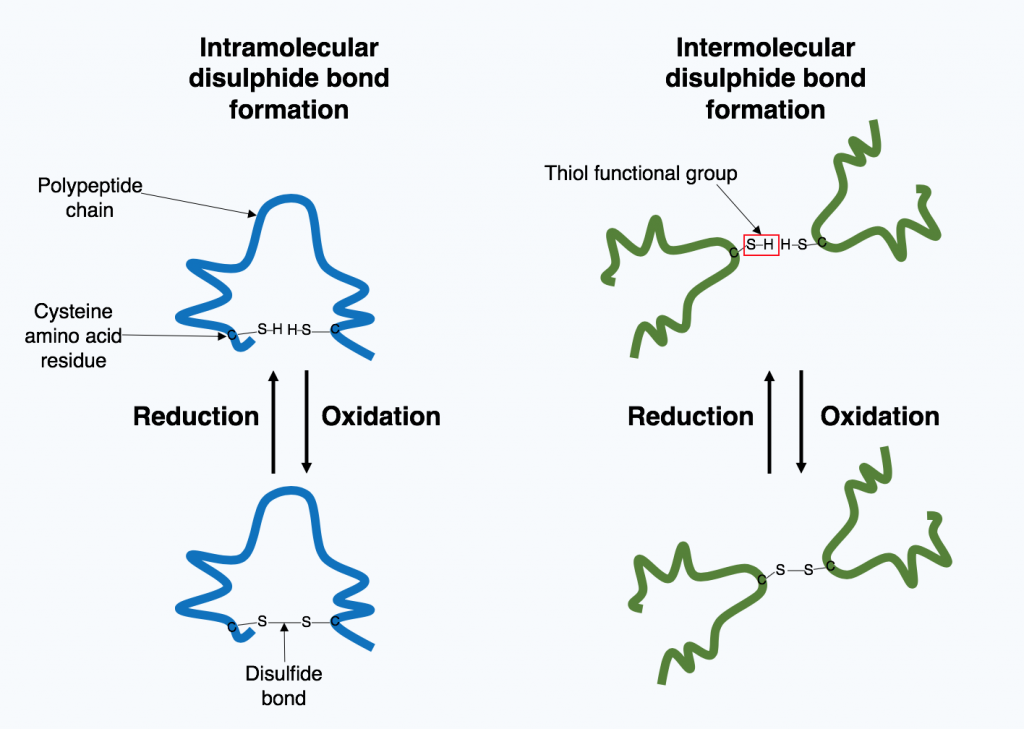
The formation of this bond depends on the environment. Oxidizing environment favors bond formation. Reducing environments break disulfide bonds.
Did you know?
1.3.4 Quaternary Structure
The fourth level of protein structure is that of quaternary structure. It refers to structures that arise as a result of interactions between multiple polypeptides, often referred to as subunits.
The subunits can be identical to each other or can be different polypeptide chains. The stabilizing interactions that hold the multiple polypeptides together are the same non-covalent interaction interactions (hydrogen bonding, ionic bonding, and hydrophobic interactions) and covalent disulfide bonds, that stabilize the tertiary structure, except, that the interactions occur between the different polypeptide chains.
There are many examples of proteins that require more than one polypeptide to be functional and we will encounter several molecular machines (DNA polymerase, RNA polymerase) that are comprised of more than 10 different polypeptides.
The nomenclature used for such multi-subunit proteins reflects the number of polypeptides and the similarity between them.
For example, Estrogen (the hormone) receptor consists of two identical peptide chains coming together to form a functional protein. This is called a homodimer. [ homo (similar) di (two) mer]
Adult hemoglobin is composed of two identical chains called α and two identical chains called β. Proteins with multiple polypeptides where at least one of the polypeptides is different from the other are Hetero-mers. Hemoglobin would be a hetero-tetra– mer.
Some clarifications of confusing terminology you may encounter!
Practice
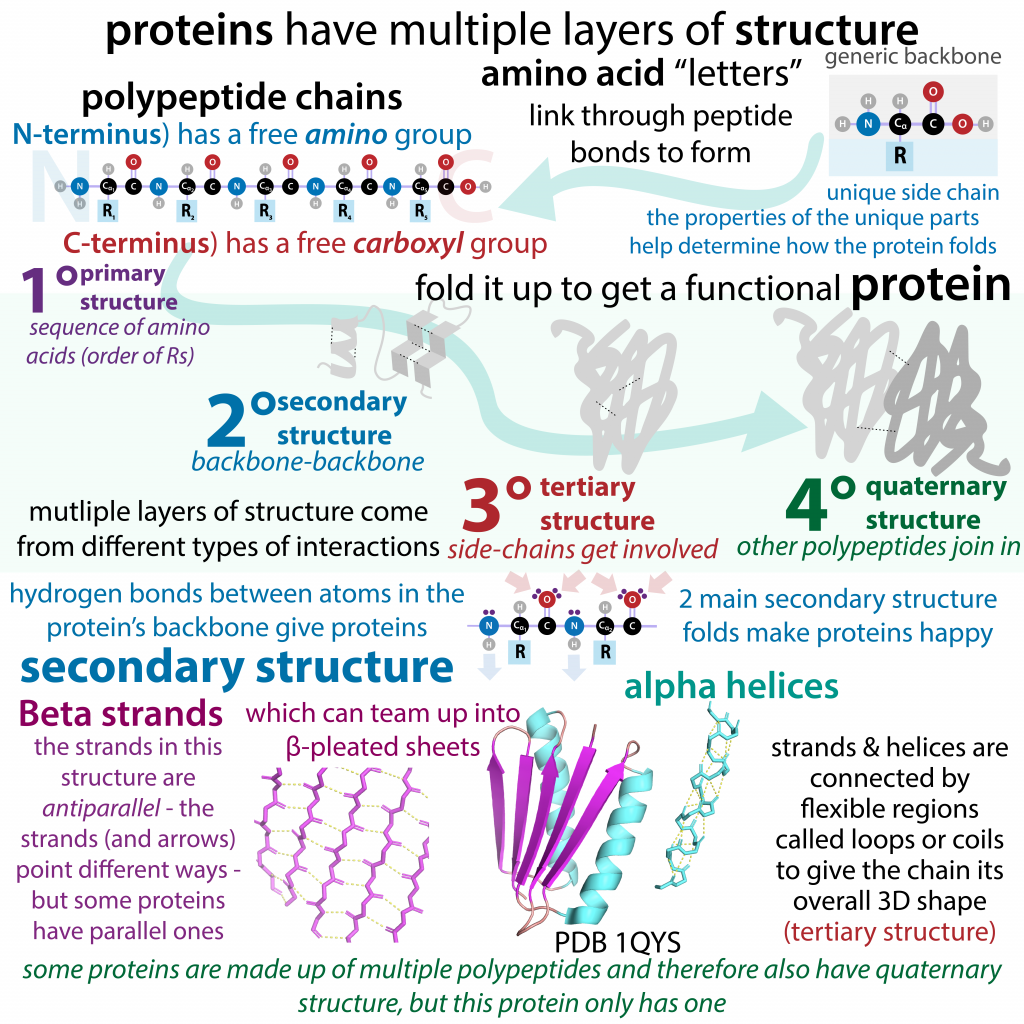
Key Takeaways: Via Sketchnotes!
A great learning tool is to create concept maps that highlight the key terms and connect them to one another in meaningful ways.
Included is an example of a free-to-use sketch note or graphic organizer uploaded by a young scientist who blogs and writes about Biochemistry. [You can find her delightful work here: “the bumbling biochemist”:https://thebumblingbiochemist.com/].
Making one for yourself makes for better learning than memorizing one. You don’t have to be an artist to do this!
Image attribution: Biochemlife, CC BY-SA 4.0 <https://creativecommons.org/licenses/by-sa/4.0>, via Wikimedia Commons
Before you continue
You should watch the Key Takeaways Lecture video linked in CANVAS!
1.4 Protein Structure Deep Dive
Features of Alpha-Helices and Beta Sheets
Shown below is a diagram for the alpha-helix highlighting the key features of the helix. (Figure 1.10)
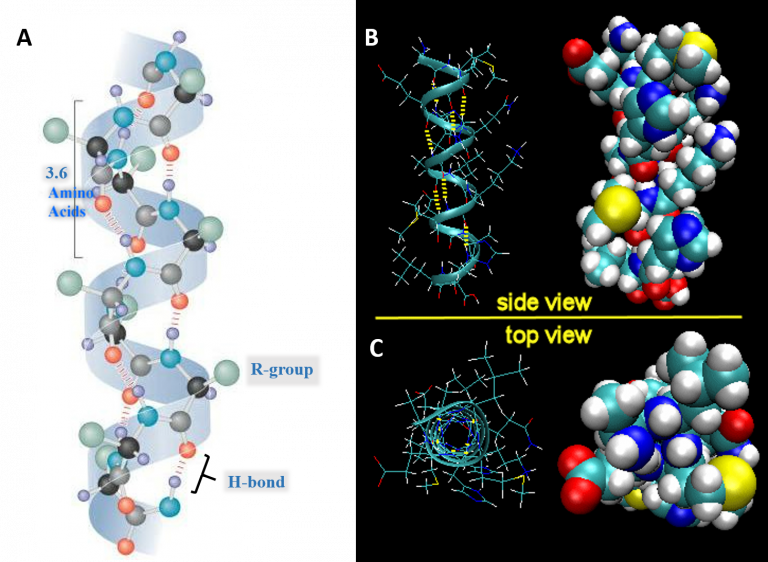
- Each full turn of the helix (360°) is 3.6 amino acids in length.
[Note that the H-bond forms between C=O groups and N-H groups in the polypeptide backbone that are four amino acids distant]
- Helices are predominantly right-handed.
- The hydrogen bonds are parallel to the axis of the helix, located inside the helix, and are in a regular arrangement. [All C=O bonds point in one direction, all N-H bonds point in opposite direction].
- R-groups extend away from the helix.
The stability of an alpha helix is affected by different factors.
- Amino acids whose R-groups are too large (tryptophan, tyrosine) or too small (glycine) destabilize α-helices.
- Glycine has a lot of conformational flexibility given its side chain is simply an H atom! Glycine is found in the more flexible regions of proteins which are the loops.
- Certain combinations of adjacent amino acids. For example, a run of positively charged or negatively charged side chains will repel one another.
- Presence of Proline- the ‘helix breaker’.
- Take a look again at the side chain of the proline in Figure 1.2.
- Notice that the hydrogen connected to the N of the amine group is not available to hydrogen bond! In fact, proline disrupts both types of secondary structures. However, proline is commonly found in turns or loops between the beta-strands, connecting secondary structure elements, and occasionally in the first helical turn where the side chain geometry does not create a problem.
Parallel and Antiparallel β-Sheets
In a β-sheet, two or more sections of the polypeptide run alongside each other and are linked in a regular manner by hydrogen bonds between the main chain C=O and N-H groups.
Take a look at the orientation of the strands in the image below. In parallel beta sheets, the arrows indicating the N-C direction are the same, where as in antiparallel sheets adjacent strands are in opposite directions. Notice how the H- bonds are angled or slightly bent in the case of parallel beta sheets, versus the more straight bond in between adjacent strand in antiparallel sheet. Image Credit: By Mysterioso – Own work, CC BY-SA 3.0, https://commons.wikimedia.org/w/index.php?curid=27351709
Image Credit: By Mysterioso – Own work, CC BY-SA 3.0, https://commons.wikimedia.org/w/index.php?curid=27351709
Practice Exercises
1.5 Domains in Protein Structure.
An important concept in protein structure is that of the protein domain. In many cases, a single polypeptide can be seen to contain two or more physically distinct substructures, known as domains. Often linked by a flexible hinge region these domains are compact, stable, and fold independently of the rest of the protein chain. Again, since the shape is important for function, domains have a shape that is best suited for a particular cellular function and are named accordingly!
For example; A nucleotide-binding domain, Calcium-binding domain. Occasionally they are named after their discoverers like Pleckstrin Homology (PH) domain. Figure 1. 10 shows the structures of two different proteins both of which have the PH domain.
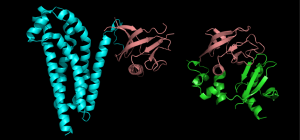
Proteins that have this domain, can bind a molecule of phosphatidyl-inositol triphosphate that is generated as part of a common cell-signaling pathway.
Proteins sharing more than a few common domains are encoded by members of evolutionarily related genes comprising gene families. Genes for proteins that share only one or a few domains may belong to gene superfamilies. Superfamily members can have one function in common (as predicted by the domain), but the rest of their sequences are otherwise unrelated.
Out of thousands of structures that have now been solved we can see similar structures among proteins with very different sequences. This suggests that there are a limited number of stable folds and almost all novelty in protein structure comes from the way these single domains are arranged. Unlike the number of novel single domains, the number of multidomain families being added to public databases is still rapidly increasing.
There are several advantages conferred by multidomain protein architecture:
- Creation of catalytic or substrate-binding sites: These sites are often formed at the interface between two domains, typically a cleft. The movement of the domains relative to each other allows the substrate to bind.
- Segregation of function.
- Multifunctional proteins A multidomain protein may have more than one function, often related, and each function is performed by a distinct domain. For example, E. coli DNA polymerase a multi-subunit protein we will encounter has both polymerase activity, and two kinds of exonuclease activity, all of which are required for DNA replication and all of which reside on distinct domains of this protein.
1.6 Protein Modifications can affect structure and function
Proteins in the cell are often modified post-translationally by the addition of functional groups via covalent bonds to the side chains of amino acids. These groups are added by enzymes. Think of these modifications as an additional makeover or accessorizing the protein. We have spent time revealing how the nature of the amino acid is crucial for protein folding, and the final shape is important for function, modifying amino acids chemically can impact the structure and the function. The changes in shape are often subtle but can have a dramatic impact on function, activity, stability, localization, and/or interacting partner molecules.
Two modifications that we will encounter later and are common in biology include:
- Addition of phosphate groups (phosphorylation)
- Addition of acetyl and methyl groups (acetylation, methylation)
Other types of modifications are shown in the diagram below (Figure 1.11).
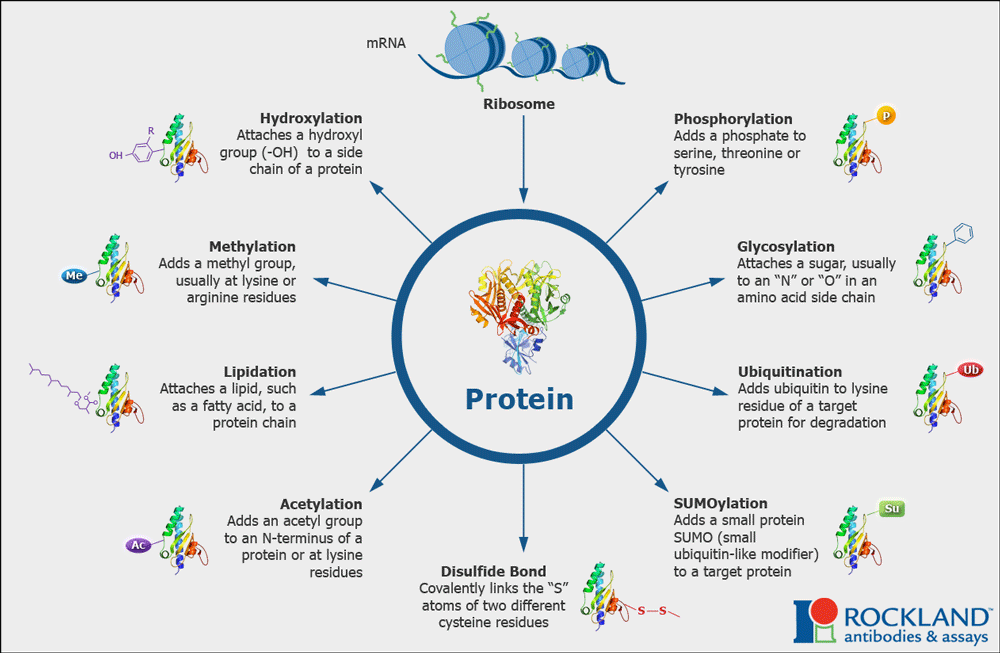
Collectively these modifications account for and enhance the molecular and functional diversity of proteins within and across species.
Much of the work of proteins inside cells involved signaling or communication. Often these modifications are reversible! Meaning that just as there are enzymes that add these groups, there are enzymes that can remove them. Having reversible modifications allows many proteins to function as signaling molecules: turning on (when modified) or off (when modifications are removed) or vice versa!
1.7 Intrinsically Disordered Proteins
We have thus far focused on how proteins can adopt a very specific three-dimensional shape. However, we now know that entire classes of proteins termed Intrinsically Disordered Proteins lack any well‐defined secondary or tertiary structure! Some proteins exhibit regions that remain unfolded (IDP regions) even as the rest of the polypeptide folds into a structured form- provided the protein “hinges” that can move a protein domain in a controlled way, loops that have an open or closed conformation.
Initially regarded as an anomaly, the presence of intrinsically disordered proteins appears to be widespread among eukaryotic proteins, it has been recognized that the observed disorder is a “feature, not a bug”.
A comparison of IDPs shows that they share sequence characteristics that appear to favor their disordered state. That is, just as some amino acid sequences may favor the folding of a polypeptide into a particular structure, the amino acid sequences of IDPs favor their remaining unfolded. IDP regions are seen to be low in hydrophobic residues and unusually rich in polar residues and proline.
The presence of a large number of charged amino acids in the IDPs can inhibit folding through charge repulsion, while the lack of hydrophobic residues makes it difficult to form a stable hydrophobic core, and proline discourages the formation of helical structures. The observed differences between amino acid sequences in IDPs and structured proteins have been used to design algorithms to predict whether a given amino acid sequence will be disordered.
What is the significance of intrinsically disordered proteins or regions? The fact that this property is encoded in their amino acid sequences suggests that their disorder may be linked to their function. The flexible, mobile nature of some IDP regions may play a crucial role in their function, permitting a transition to a folded structure upon binding a protein partner or undergoing post-translational modification.
Studies on several well-known proteins with IDP regions suggest some answers. IDP regions may enhance the ability of proteins like the lac repressor to translocate along the DNA to search for specific binding sites. The flexibility of IDPs can also be an asset in protein-protein interactions, especially for proteins that are known to interact with many different protein partners.
1.8 X-Ray Crystallography: Art Marries Science
How would you examine the shape of some thing too small to see in even the most powerful microscope?
Scientists trying to visualize the complex arrangement of atoms within molecules have exactly that problem, so they solve it indirectly. By using a large collection of identical molecules — often proteins — along with specialized equipment and computer modeling techniques, scientists are able to calculate what an isolated molecule would look like.
The two most common methods (prior to computational methods of protein prediction) used to investigate molecular structures are X-ray crystallography (also called X-ray diffraction) and nuclear magnetic resonance (NMR) spectroscopy. Researchers using X-ray crystallography grow solid crystals of the molecules they study. Those using NMR study molecules in solution. Each technique has advantages and disadvantages. Together, they provide researchers with a precious glimpse into the structures of life.
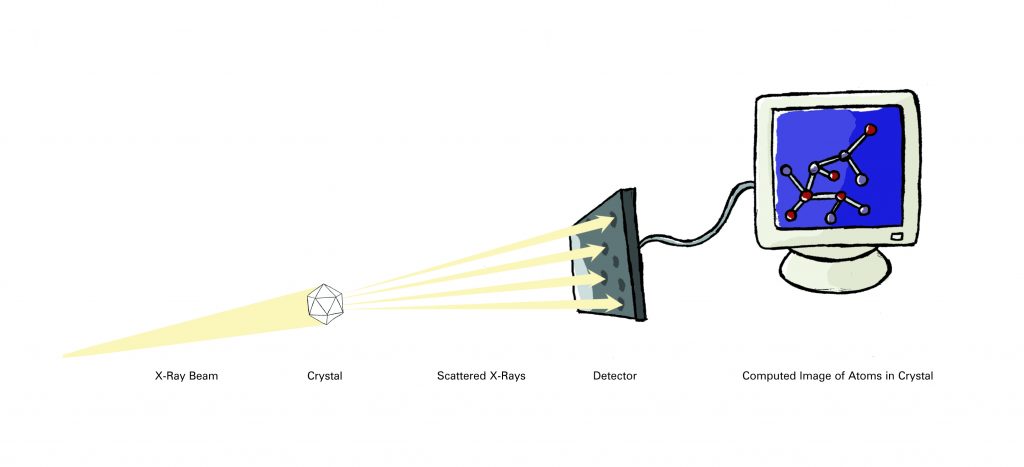
More than 85 percent of the protein structures that are known have been determined using X-ray crystallography. In essence, crystallographers aim high-powered X-rays at a tiny crystal containing trillions of identical molecules. The crystal scatters the X-rays onto an electronic detector like a disco ball spraying light across a dance floor. The electronic detector is the same type used to capture images in a digital camera. After each blast of X-rays, lasting from a few seconds to several hours, the researchers precisely rotate the crystal by entering its desired orientation into the computer that controls the X-ray apparatus. This enables the scientists to capture in three dimensions how the crystal scatters, or diffracts, X-rays. The intensity of each diffracted ray is fed into a computer, which uses a mathematical equation called a Fourier transform to calculate the position of every atom in the crystallized molecule.
The result — the researchers’ masterpiece — is a three-dimensional digital image of the molecule. This image represents the physical and chemical properties of the substance and can be studied in intimate, atom-by-atom detail using sophisticated computer graphics software.
Crystal Cookery
An essential step in X-ray crystallography is growing high-quality crystals. The best crystals are pure, perfectly symmetrical, three-dimensional repeating arrays of precisely packed molecules. They can be different shapes, from perfect cubes to long needles. Most crystals used for these studies are barely visible (less than 1 millimeter on a side). But the larger the crystal, the more accurate the data and the more easily scientists can solve the structure.
Crystallographers grow their tiny crystals in plastic dishes. They usually start with a highly concentrated solution containing the molecule. They then mix this solution with a variety of specially prepared liquids to form tiny droplets (1-10 microliters). Each droplet is kept in a separate plastic dish or well. As the liquid evaporates, the molecules in the solution become progressively more concentrated. During this process, the molecules arrange into a precise, three-dimensional pattern and eventually into a crystal — if the researcher is lucky.
Sometimes, crystals require months or even years to grow. The conditions — temperature, pH (acidity or alkalinity), and concentration — must be perfect. And each type of molecule is different, requiring scientists to tease out new crystallization conditions for every new sample. Even then, some molecules just won’t cooperate. They may have floppy sections that wriggle around too much to be arranged neatly into a crystal. Or, particularly in the case of proteins that are normally embedded in oily cell membranes, the molecule may fail to completely dissolve in the solution.
Some crystallographers keep their growing crystals in air-locked chambers, to prevent any misdirected breath from disrupting the tiny crystals. Others insist on an environment free of vibrations — in at least one case, from rock-and-roll music. Still, others joke about the phases of the moon and supernatural phenomena. As the jesting suggests, growing crystals remains one of the most difficult and least predictable parts of X-ray crystallography.
It’s what blends art with science.
Watch the lecture videos in this playlist: Understanding X-ray crystallography
SOME KEY TERMS
References and Attributions
This chapter is curated from and contains material from the following CC-licensed content. Changes include rewording, replacing, and removing paragraphs with original material.
- Section 1.1 and Section 1.8 from Structure of Life (2007). Booklet, retrieved from National Institute of General Medical Sciences https://www.nigms.nih.gov/education/Booklets/The-Structures-of-Life/Pages/Home.aspx.; (United States Government Work, in the public domain)
- Ahern K, Rajagopal I and Tan T. (2013). Biochemistry Free for All (Version 1.3). Licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. The entire textbook is available for free from the authors at http://biochem.science.oregonstate.edu/content/biochemistry-free-and-easy.
- Details of Protein Structure” by Gerald Bergstrom, LibreTexts which is licensed under CC BY. The chapter can be found online at https://bio.libretexts.org/@go/page/16428
Images
Unless otherwise noted within the text, images on this page are licensed under CC-BY 4.0 by OpenStax. Located at: https://openstax.org/books/biology/pages/3-4-proteins.Access for free at https://openstax.org/books/biology/pages/1-introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}