
Tools and Techniques of Molecular Biology
Recombinant DNA Technology
Learning Objectives
When you have mastered the information in this chapter, you should be able to:
- Describe the characteristics of plasmids/ cloning vectors
- List in words or indicate in a drawing the important features of a plasmid vector that are required to clone a gene. Explain the purpose of each feature.
- Explain how vectors are used in cloning a gene.
- Explain what are expression vectors and how they are used.
- Explain how to use restriction enzymes to create a recombinant plasmid.
- What properties of the DNA restriction fragments enable ligation of these fragments?
- Explain what role do restriction enzymes have in nature?
- Explain how DNA ligase is used to create a recombinant plasmid
- Describe possible recombinant plasmids that form when ligating a restriction digest
- Design a cloning experiment/strategy when given vector sequences and insert sequences
- Predict or troubleshoot the reasons for a ‘failed’ cloning experiment.
- Due to a mishap in the lab, bacteria carrying a plasmid with a kanamycin-resistant gene and bacteria carrying a plasmid with an ampicillin-resistance gene were accidentally mixed together. How would you design an experiment allowing you to sort out the two kinds of bacteria?
- Explain the use of reporter gene assays.
- Outline an experiment using reporter gene assays to identify regulatory elements in promoter sequences of genes (prokaryotic and eukaryotic)
Introduction
It is likely you know somebody who has diabetes (a disease that occurs when a person’s blood glucose [sugar] is too high). In certain types of diabetes, the body is unable to manufacture or produce insulin, the protein hormone needed to manage blood glucose levels. In Chapter 1 and Chapter 2 you learned about the breakthrough research that led to the isolation of insulin, first from dogs and subsequently from pancreases of cows and subsequent demonstration that purified insulin when injected in humans was able to treat the symptoms.
By 1923, insulin had become widely available in mass production, and Banting and Macleod were awarded the Nobel Prize in medicine. Diabetes was no longer a life-shortening disease. While these methods were effective, using animal-produced proteins sometimes resulted in adverse reactions, and making large enough quantities was difficult. This all changed with the advent of recombinant DNA technology.
This technology allowed scientists to take human DNA (for example insulin gene) and introduce it into bacterial DNA, allowing the bacteria to produce a human protein.
The ability to make recombinant DNA is such a seminal technology that just realizing it could be done and then doing it in a test tube for the first time earned Paul berg a half-share in the 1980 Nobel Prize in Chemistry (the other half was shared by Walter Gilbert and Frederick Sanger for studies that enabled efficient DNA sequencing).
Molecular Cloning and Recombinant DNA Technology
Joining together of DNA fragments from different sources creates recombinant DNA. The ability to cut and paste DNA might seem like purely a technical feat, but one key application that arose out of this is molecular cloning.
Typically cloning is referring to ‘molecular cloning’, the process of making multiple copies of a particular sequence of DNA, expression of genes, and study of specific genes.
In molecular cloning a gene of interest can be inserted into a vector, usually a plasmid, by cutting both the vector and the gene (called the insert) with the same enzyme to generate sticky ends and joining the two pieces together to generate a recombinant.
A plasmid is a type of autonomously replicating, extrachromosomal DNA. It is quite simple to extract plasmids from the cells, engineer them to contain the gene of interest and re-introduce the recombinant plasmid into the bacteria. The idea was that when the plasmid DNA was replicated, the extra inserted gene would also be copied. Thus, by growing up a lot of the bacteria carrying the plasmid, many copies of the gene of interest could be obtained, to provide sufficient amounts of the gene to use in experiments.
How do you begin to ‘clone’ a gene or make a recombinant DNA molecule?
First, let’s think about what ‘cloning’ requires conceptually.
- We need to isolate the gene/ DNA sequence (for example, a gene for a human therapeutic protein) away from the rest of the genome. This would involve cutting DNA in a reproducible manner and procedures for viewing the DNA fragments.
- We need a way to ‘paste’ this DNA into a vector (a different DNA molecule) to create a recombinant molecule
- This vector should be able to replicate inside a host cell.
- We need procedures for introducing the recombinant DNA molecule into the appropriate cell (eukaryotic or prokaryotic)
Key developments that allowed for the revolution in recombinant DNA technology came from studying prokaryotic biology.
These include the identification of DNA cutting enzymes “restriction endonucleases”, isolation of small extrachromosomal DNA found in bacteria called ‘Plasmids’ and knowledge that bacteria can take up DNA from exogenous sources- ‘transformation’.
Digestion of DNA with restriction endonucleases, pasting with ligases
The creation of recombinant DNA molecules is possible due to the use of naturally occurring restriction endonucleases (restriction enzymes), bacterial enzymes produce as a protection mechanism to cut and destroy foreign cytoplasmic DNA that is most commonly a result of bacteriophage infection. Stewart Linn and Werner Arber discovered restriction enzymes in their 1960s studies of how E. coli limits bacteriophage replication or infection (Read Link to learning).
Today, we use restriction enzymes extensively for cutting DNA fragments that can then be pasted with another DNA molecule to form recombinant molecules. These enzymes are purified from various bacterial species and can be purchased commercially. They are named after the bacterium from which they were first isolated. For example, EcoRI and EcoRV are both enzymes from E. coli. HindIII came from the bacteria Haemophilus influenzae.
Restriction enzymes serve as molecular scissors recognizing a specific sequence in the DNA called restriction sites. The sites are usually palindromic, typically between four to six base pairs in length.
Palindromic sequences are the same sequence forwards and backward. Some examples of palindromes: RACE CAR, CIVIC, A MAN A PLAN A CANAL PANAMA.
With respect to DNA, there are 2 strands that run antiparallel to each other. Therefore, the reverse complement of one strand is identical to the other. This means that the sequence of the recognition site when read in the 5‘ to 3‘ direction for the top strand is exactly the same as that of the bottom strand. Upon binding this sequence, the enzyme cuts both strands of the DNA generating a double-stranded break.
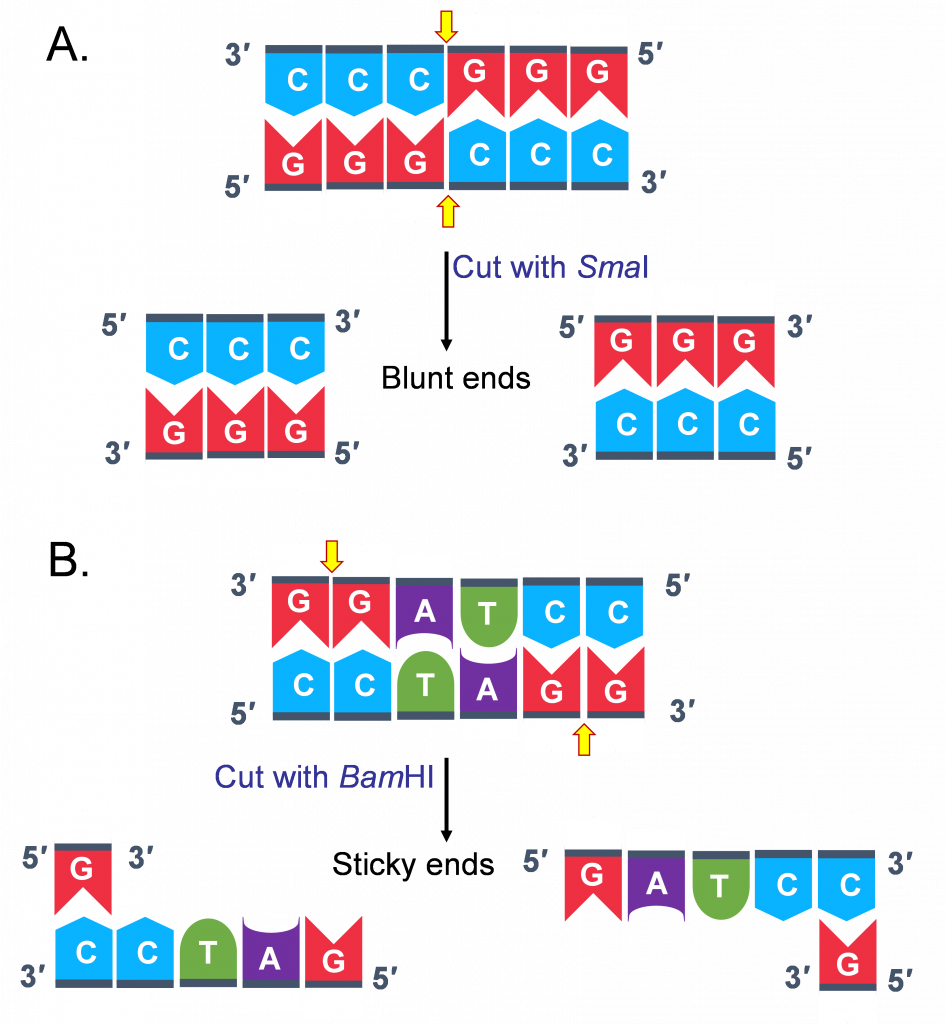
However, based on where the enzyme cuts different kinds of ends are generated. Some enzymes cut asymmetrically leaving a overhang at the 5’ end of one strand of the duplex; some leave an overhang at the 3’ end. These ends are referred to as “sticky ends”. (Figure 2B)
Some enzymes cut symmetrically leaving no overhanging sequence overhangs and generate “blunt ends”(Figure 2A)
This distinction in cutting is important because “sticky ends” are thus called because they are self-complementary!
Molecules with complementary or compatible sticky ends can easily anneal (or stick to one another) forming hydrogen bonds between complementary bases, at their overhangs.
With the help of an enzyme we are already familiar with, DNA Ligase the 2 pieces of DNA are joined together through covalent bonding, making the molecule a continuous double strand.
With a combination of RE and DNA ligase, any two fragments regardless of their origin (animal, plant, fungal, bacterial) can be joined in vitro to form recombinant molecules, especially if they have compatible sticky ends.

Lecture Activities
3. Go through this scrollable interactive on LabExchange. It demonstrates how restriction enzymes work and why they can be used as tools to analyze DNA and create recombinant plasmids.
TRY THIS ACTIVITY:
Link to Learning: Discovery of Restriction Endonucleases.
Why would bacteria have enzymes that could cut up DNA?
The discovery of this special class of enzymes came from early work in the 1950s on certain strains of bacteria. Scientists observed that certain strains of E. coli, a common bacterium found in the human gut, were resistant to infection by bacteriophage—a virus that infects bacteria by injecting its DNA into the cell and commandeering the host cell’s molecular processes to make more bacteriophage. This peculiar (at the time) phenomenon was called ‘Host-Restriction”. 1960s, Werner Arber was investigating the possible mechanisms and observed a dramatic change in the bacteriophage DNA after it invaded these resistant strains of bacteria: the bacteriophage DNA was degraded and cut into pieces! To explain the resistance of certain bacterial strains to bacteriophage infection, Arber hypothesized that bacteriophage-resistant bacterial cells might express a specific enzyme that degrades only invading bacteriophage DNA, but not their own DNA. Further investigation of this primitive bacterial “immune system” led to the discovery of restriction enzymes, proteins that recognize cut DNA at specific sequences called restriction sites. Thus, they restrict the growth of bacteriophages by recognizing and destroying the phage DNA. Read the following article to learn how bacteria protect their own DNA from being chewed up.
(https://www.nature.com/scitable/spotlight/restriction-enzymes-18458113/)
Plasmids
Bacteria carry a single circular genome (chromosome) that carries all the information needed by the organism to survive and reproduce. However, in addition, many bacteria also carry small circular DNA molecules, ranging in size from 1,000 to 200,000 base pairs present in multiple copies separate from the chromosomal DNA called Plasmids.
Plasmids carry some useful features:
- They have their own origin of replication, which allows the plasmid to produce many copies (replicate) inside of a bacterial cell. Additionally, as mentioned above they replicate at a higher frequency than the bacterial chromosome, which means there could be thousands of copies of plasmid within a single cell.
- They have a promoter for transcription of an inserted gene if so desired.
- They carry a gene for antibiotic resistance, that provides for a mechanism to select for a small percentage of bacterial cells that have taken up the plasmid
Scientists have taken advantage of plasmids to use them as tools to clone, transfer, and manipulate genes. Plasmids that are used experimentally have been repurposed to make them suitable for cloning and called vectors.
Often the terms plasmid and vector are used interchangeably.
Vectors (plasmids) used by scientists today come in many sizes and vary broadly in their functionality. The greatest variety of cloning vectors has been developed for use in the bacterial host E. coli.
They all however must have at least three features
1) An origin of replication
2) A selectable marker (antibiotic resistance gene)
Cells that take up the plasmid will be able to grow in the presence of the antibiotic. If bacterial cells to which the plasmid has been added are plated on agar containing the antibiotic, the cells which took up the plasmid will be able to grow, while the others will not.
3) A cluster of restriction enzyme sites which called a Multiple cloning site (MCS).
The MCS enables researchers to introduce any piece of DNA into the vector by restriction enzymes digestion and ligation. As a general rule, the restriction sites in the MCS are unique and not located elsewhere in the plasmid backbone, which is why they can be used for cloning by restriction enzyme digestion but when designing a cloning experiment a detailed ‘Restriction Map’ can be obtained”.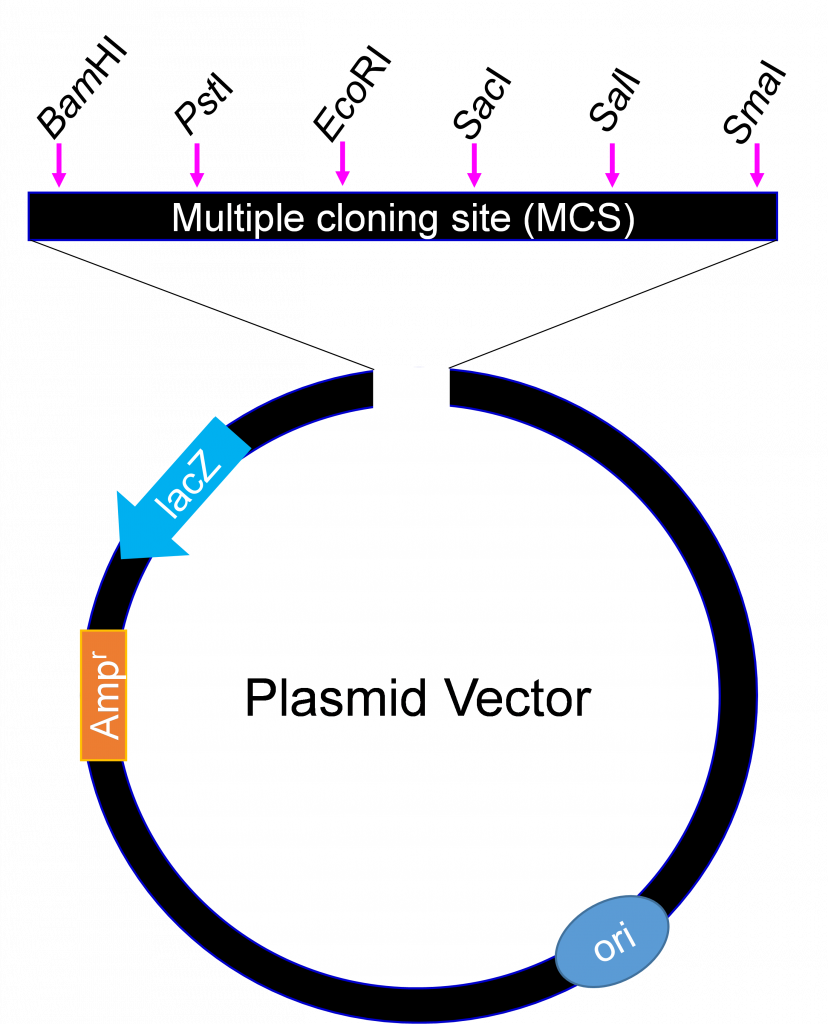
Watch this video embedded below or go to this Youtube Link from Addgene
Building a recombinant plasmid: The workflow
Below is the general workflow of how molecular cloning takes place.
Step 1. Isolate DNA of interest (called insert in a cloning experiment)
Step 2. Identify cloning vector/plasmid
Step 3. – once the target and vector DNA have been identified, both types of DNA are cut using restriction endonucleases.
Step 4. Purify the cut fragments: U
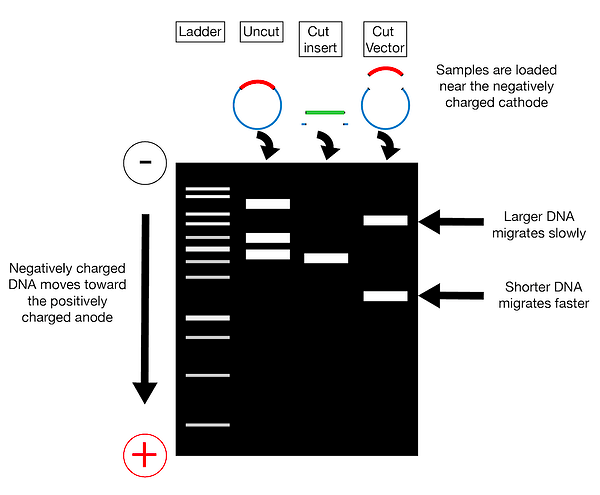
Step 4. Combine purified target and vector DNA – the two types of DNA are combined together in a single tube with the addition of DNA ligase and appropriate buffer. This results in the creation of recombinant DNA.
Step 5. Introduce recombined molecule into host cell
Once the target DNA has been stably combined with vector DNA, the recombinant DNA must be introduced into a host cell, in order for the genes to be replicated or expressed. There are different methods for introducing the recombinant DNA, largely depending upon the complexity of the host organism.
In the case of bacteria, transformation is often the easiest method, using competent cells to pick up the recombinant DNA molecules. Alternatively, electroporation can be used, where the cells are exposed to a brief pulse of high–voltage electricity causing the plasma membrane to become temporarily permeable to DNA passage.
Step 6. Grow the bacteria and plate on agar plates that contain the antibiotic. Bacterial cells carrying the plasmid will grow, multiply and form visible colonies.
The steps involved in molecular cloning using bacterial transformation are outlined in this graphic flowchart.

Cloning with Restriction Enzymes- Some Considerations
When designing a cloning experiment where you want to cut out an insert from one source such as genomic DNA and insert it into a second source such as a plasmid (vector), there are a few considerations.
1) The restriction sites should occur in the desired location in your recipient plasmid (usually in the Multiple Cloning Site (MCS) but not elsewhere in the vector.
2) They should be on either side of your insert, but do not cut within your insert!
3) Using 2 different enzymes makes cloning more efficient preventing the plasmid from reforming a circle without the inserted gene. Importantly it allows the inserted gene to be oriented in the correct position for transcribing the “sense” strand of DNA (the strand that codes for the protein)!
As we learned in Introduction to Transcription, the strand of DNA that has the coding sequence of the gene (5′-3′ direction) is called the Sense strand. The correct orientation in the recipient plasmid is key- you don’t want to express the antisense version of your gene!
See an example where I walk through the logic of picking restriction enzymes to use.
Cloning by PCR
While we mostly focused on restriction enzyme cloning. Another convenient way to isolate DNA sequence of interest is Polymerase chain reaction (PCR)! The basic steps in PCR reaction were discussed earlier.
This by far the most common method of cloning. In this method when designing primers for your region of interest researchers in introduce restriction site to the 5′ end of the primers.
Then upon amplification the PCR product will have a restriction site added to either end that can be digested with the appropriate restriction enzyme. If the other primer has a different restriction sequence then the PCR fragment can be inserted in a directional dependent manner in a host plasmid.
Types of Plasmids/Vectors
There are many different types of plasmids or vectors for use in molecular biology experiments. The choice of vector depends on the biological question or ultimate use of the cloned DNA. Ones you will encounter in this course or are
General Purpose Vectors: Used to facilitate the cloning of DNA fragments. Cloning vectors tend to be very simple, often containing only a bacterial resistance gene, origin of replication, and MCS. They are small and optimized to help in the initial cloning of a DNA fragment.
Genome Engineering Plasmids: Used for editing and modifying genes using CRISPR, which we shall see in Genome Editing chapter.
Expression Vectors: Used for gene expression (for the purposes of gene study). Expression vectors must contain a promoter sequence, a transcription terminator sequence, and the inserted gene. The promoter region is required for the generation of RNA from the insert DNA via transcription. The terminator sequence on the newly synthesized RNA signals for the transcription process to stop.
Many sophisticated variations on such vectors have been created that have made it easy to produce and purify large amounts of any protein of interest for which the gene has been cloned.
For example: A handy feature in some expression vectors is a sequence encoding an affinity tag either up- or downstream of the gene being expressed. This sequence allows a short affinity tag (such as a run of histidine residues) to be fused onto the encoded protein. The tag can be used to readily purify the protein, as described in the section on affinity chromatography using special beads that bind to His-tag.
Another handy feature is fusing the coding sequence of fluorescent proteins like GFP to the coding sequence of gene of interest and create a recombinant fusion protein. This allows researchers to view the presence of the protein inside a living cell by microscopy. The two genes are under the same promoter element, transcribed as though it is one gene into a single messenger RNA molecule. The mRNA is then translated into protein.
To connect back to protein folding, in these cases, it is important that both proteins be able to properly fold into their active conformations when fused together and interact with their substrates despite being fused. GFP protein is small and is a single domain protein. It has been shown to adopt its three- dimensional shape independent of the addition of extra amino acids in many cases. Creating fluorescent fusion proteins is a very important tool used by cell biologist (see side bar link to learning for stunning live cell microscopy of a 10 day tumor metastasizing)
To see a 10-day time-lapse of an engineered human micro-tumor forming and metastasizing click here.
More at: https://www.nikonsmallworld.com/galleries/2021-small-world-in-motion-competition
In all examples of Expression Cloning, where the goal is to transcribe mRNA and/or also have the host organism (Bacterial or Eukaryotic) make protein cloning; directionality matters!
Another special type of expression vector is a reporter plasmid which we discuss in detail below.
Reporter Plasmids and Reporter Gene Assays: Tool to Study Gene Expression
A common tool for studying the regulation of gene expression by cis- or trans-acting factors, are reporter gene assays.
In these assays, a ‘reporter gene’ acts as a surrogate (reports) for the coding region of the gene under study. Commonly used reporter genes are those that have visually identifiable characteristics usually involve fluorescent and luminescent proteins.
Examples include the gene that encodes jellyfish green fluorescent protein (GFP), which causes cells that express it to glow green under blue light, the enzyme luciferase, which catalyzes a reaction with luciferin to produce light, a
Other genes that serve as good reporters are types of enzymes that themselves may not be visible when made, but upon addition of a substrate generate a color that can be measured.
A common example of such a gene is Lac Z from E.coli which encodes β-galactosidase. This enzyme causes bacteria expressing the gene to appear blue when grown on a medium that contains the substrate analog X-gal or cells that express the gene to stain blue. The choice of reporter gene will often depend on biological question.
Using recombinant DNA technology one or more gene regulatory elements being analyzed are introduced (cloned) upstream of the coding sequence of the reporter gene. Following introduction of the reporter construct into cells and experimental treatment, expression levels of the reporter gene are monitored by quantifying the reporter protein enzymatic activity.
References and Attributions
This chapter contains material taken from the following CC-licensed content. Changes include rewording, removing paragraphs and replacing with original material, and combining material from the sources.
1. Bergtrom, Gerald, “Cell and Molecular Biology 4e: What We Know and How We Found Out” (2020). Cell and Molecular Biology 4e: What We Know and How We Found Out – All Versions. 13.
https://dc.uwm.edu/biosci_facbooks_bergtrom/13
2. Works contributed to LibreTexts by Kevin Ahern and Indira Rajagopal. LibreTexts content is licensed by CC BY-NC-SA 3.0. The entire textbook is available for free from the authors at http://biochem.science.oregonstate.edu/content/biochemistry-free-and-easy
3. Genetics, Agriculture, and Biotechnology by Walter Suza; Donald Lee; Marjorie Hanneman; and Patricia Hain is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License
4. OpenStax Microbiology. Provided by: OpenStax CNX. Located at: http://cnx.org/contents/e42bd376-624b-4c0f-972f-e0c57998e765@4.2. License: CC BY: Attribution. License Terms: Download for free at http://cnx.org/contents/e42bd376-624b-4c0f-972f-e0c57998e765@4.2
